

制药行业专家经验分享回顾以及现场问答总结 | 加速产品无菌检测与放行——快速无菌检测法规更新与中外实践

导读 10月17日晚八点,梅里埃工业技术中心行业技术专家(健康护理行业) 赵思扬老师做客医麦课堂:从当前传统无菌检测出发,就传统方法的不足引申出快速检测方法,并系统梳理了中外法规的相关章节,实践分析,展示了快速无菌检测方法如何助力企业加速产品无菌检测与放行。
01 | 无菌检查法 |
无菌检查是用于检查药典要求无菌的药品、生物制品、医疗器具、原料辅料及其他品种是否无菌的方法。生物制品,作为药典要求的无菌药品之一,必须在放行之前完成无菌检查。
在分享中,赵老师介绍了近年来药典中无菌检查法暴露出的局限性,主要体现为:
▪ 培养基的种类、培养温度和培养时间的选定是折中妥协的结果,并非所有微生物都能在此条件下良好的生长(有超过30%无菌检查失败发生在第7-14天)
▪ 部分生物制品(如疫苗)会引起培养基变浑浊,从而影响判读,故必须进行转种
▪ 检测并记录培养基的浑浊、絮状菌膜形成和沉淀积累,完全依赖操作者主观判读
▪ 14天的培养时间无法满足药品效期的需求。
02 | 各地区短效期产品无菌检查法规 |
美国检查法规

21 CFR 211.165章节提到供应产品的测试与放行:“当放射性药物等短效期产品施行无菌或热原检查时,该批次可能需在测试完成前放行,因此需尽快完成。”
USP已施行针对CSPs、PET造影剂、细胞和基因疗法产品等的<1071>.Rapid Microbial Tests for Release of Sterile Short-life Products: A Risk-basedApproach章节。
欧洲检查法规
EP9.2版本(2017年7月)中更新了2.6.27 Microbiological Examinationof Cell-based Preparations章节,考虑到产品的输注效期、本体构成和药品接种量,引入了基于生长的自动化方法,这一法规的改动是配合FDA所批准的两例CAR-T产品进入欧洲市场。
03 | 替代方法验证 |
现阶段替代方法的应用需要经过验证后进行报批。
现行《中国药典》四部9201章节为所采用的试验方法能否替代药典规定的方法提供指导。如果出于成本、生产量、快速简便、提高质量等需要,检验方法不是药典或标准中规定的方法,使用前应进行替代方法的验证,确认其应用效果优于或等同于药典方法。
04 | 下面是USP和EP中收录的两个代表性方法 |
USP收载的固相细胞计数法:
▪ 适用于可过滤的样品检测
▪ 荧光酶活标记技术可协助区分出活的微生物细胞
▪ 可在约3.5h内获得结果
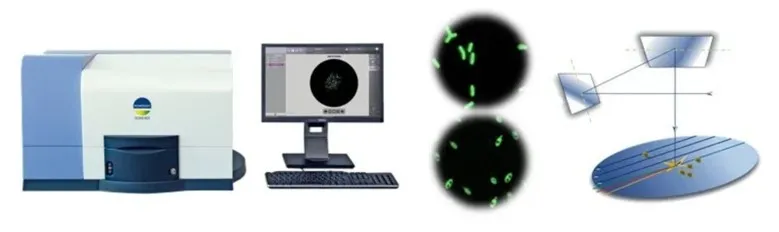
点击上方图片了解SCANRDI
USP/EP收载的呼吸法:
▪ 适用于不可过滤的样品检测
▪ 微生物代谢中产生的CO2检测,无需依赖培养基浑浊的判读
▪ 可在7天内获得结果(用户可基于风险自行验证)

 网站首页
网站首页 产品系列
产品系列